







http://glstring.org
http://loinc.org
http://unstats.un.org/unsd/methods/m49/m49.htm
http://varnomen.hgvs.org
http://www.ebi.ac.uk/ipd/imgt/hla
http://www.ncbi.nlm.nih.gov/projects/SNP
http://www.ncbi.nlm.nih.gov/refseq
http://www.nlm.nih.gov/research/umls/rxnorm
urn:ietf:bcp:47
This fragment is available on index.html
This publication includes IP covered under the following statements.
| Type | Reference | Content |
|---|---|---|
| web | example.org | http://example.org/identifiers/files/11132 |
| web | example.org | http://example.org/identifiers/files/11125 |
| web | example.org | http://example.org/identifiers/files/11111 |
| web | example.org | http://example.org/identifiers/files/11150 |
| web | www.example.com | telecom : ph: (100) 200-3000, fax: (400) 500-6000, http://www.example.com/mytestpathlabs |
| web | snomed.info | 444734003 |
| web | snomed.info | 399223003 |
| web | www.genenames.org |
Include all codes defined in
http://www.genenames.org
version Not Stated (use latest from terminology server)
|
| web | github.com | Genomics Reporting Implementation Guide, published by HL7 International / Clinical Genomics. This guide is not an authorized publication; it is the continuous build for version 4.0.0-cibuild built by the FHIR (HL7® FHIR® Standard) CI Build. This version is based on the current content of https://github.com/HL7/genomics-reporting/ and changes regularly. See the Directory of published versions |
| web | en.wikipedia.org | Secondary findings are genetic test results that provide information about variants in a gene unrelated to the primary purpose for the testing, most often discovered when Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) is performed. This extension should be used to denote when a genetic finding is being shared as a secondary finding, and ideally refer to a corresponding guideline or policy statement. |
| web | en.wikipedia.org | Secondary findings are genetic test results that provide information about variants in a gene unrelated to the primary purpose for the testing, most often discovered when Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) is performed. This extension should be used to denote when a genetic finding is being shared as a secondary finding, and ideally refer to a corresponding guideline or policy statement. |
| web | gnomad.broadinstitute.org | component:knowledge-base : indicates the database from which the annotation is derived. Annotations are obtained from different knowledge bases (e.g. Clinvar , gnomAD , 1000 Genomes ). Knowledge bases continue to evolve, with new ones being developed, and existing ones being updated/revised. |
| web | www.internationalgenome.org | component:knowledge-base : indicates the database from which the annotation is derived. Annotations are obtained from different knowledge bases (e.g. Clinvar , gnomAD , 1000 Genomes ). Knowledge bases continue to evolve, with new ones being developed, and existing ones being updated/revised. |
| web | glstring.org | For HLA, KIR, and other genes in the immunogenomics domain, the National Marrow Donor Program (NMDP) led a community effort to define the Genotype List String (GL String) grammar, described here . Notably, the GL String uses '+' as a delimiter between alleles in a genotype. It also has delimiters for ambiguous genotypes, allele lists, and haplotypes. |
| web | gnomad.broadinstitute.org |
knowledge-base (component)
indicates the database from which the annotation is derived. Annotations are obtained from different knowledge bases (e.g. Clinvar
, gnomAD
, 1000 Genomes
). Knowledge bases continue to evolve, with new ones being developed, and existing ones being updated/revised.
|
| web | www.internationalgenome.org |
knowledge-base (component)
indicates the database from which the annotation is derived. Annotations are obtained from different knowledge bases (e.g. Clinvar
, gnomAD
, 1000 Genomes
). Knowledge bases continue to evolve, with new ones being developed, and existing ones being updated/revised.
|
| web | www.oncokb.org | Oncokb levels |
| web | bioportal.bioontology.org | Bioportal clinical variant interpretation |
| web | civic.readthedocs.io | CIViC Evidence |
| web | civic.readthedocs.io | CIViC Significance |
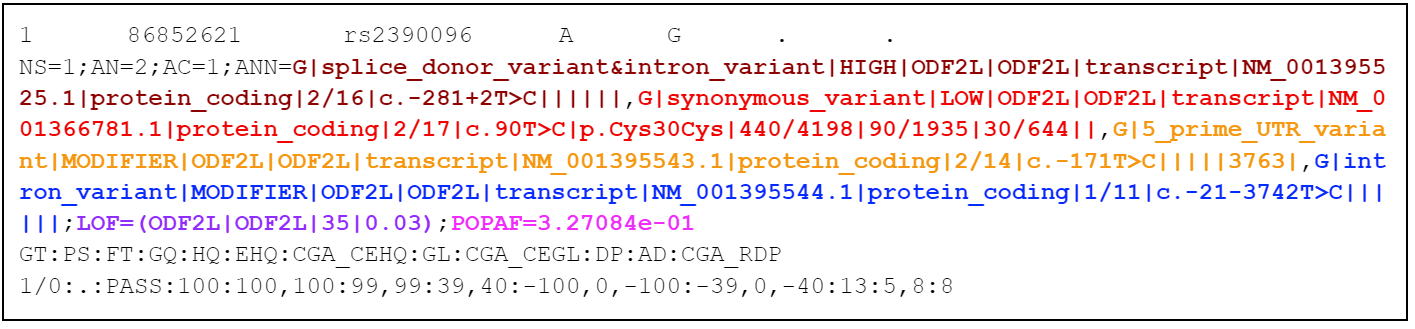
| web | example.org | This next figure shows a VCF row. A variant in the ODF2L gene has been annotated with the snpEff/snpSIFT variant prediction tool . Things to note include: |
| web | gnomad.broadinstitute.org | This example uses snpSIFT with gnomAD to annotate a population allele frequency |
| web | snomed.info | 444734003 |
| web | snomed.info | 399223003 |
| web | smarthealthit.org | What is SMART? |
| web | cancer.sanger.ac.uk | COSMIC |
| web | glstring.org | GL String |
| web | www.genenames.org | The HUGO Gene Nomenclature Committee (HGNC) table carries the gene ID, gene symbol and full gene name. Gene IDs must be sent as codes and begin with "HGNC:". Gene symbols should be sent as display. GENE IDs are specific to the species, whereas gene symbols and names are shared by all species with the same gene. The HGNC-Symb table provided by HUGO carries only human genes and is available in a table by LHC. The code for this coding system is the HGNC gene code, the "name" or print string is the HGNC gene symbol. More than 28,000 human gene symbols and names have been assigned so far, including almost all of the protein coding genes. NCBI creates what might be thought of as interim codes. Older systems might send HGNC gene IDs without the "HGNC:" prefix. This is not following the guidance of the creators of the code system, and introduces ambiguity. So caution must be taken to confirm alignment. HGNC also provides an index on gene families/groups. GeneGroup IDs do not begin with "HGNC:", so care must be made to ensure alignment of concepts when viewing an HGNC ID from an older system that may be referring to the GeneID and not a gene group. For example, 588 refers to the HLA gene family, but HGNC:588 identifies the ATG12 gene. |
| web | www.genenames.org | The HUGO Gene Nomenclature Committee (HGNC) table carries the gene ID, gene symbol and full gene name. Gene IDs must be sent as codes and begin with "HGNC:". Gene symbols should be sent as display. GENE IDs are specific to the species, whereas gene symbols and names are shared by all species with the same gene. The HGNC-Symb table provided by HUGO carries only human genes and is available in a table by LHC. The code for this coding system is the HGNC gene code, the "name" or print string is the HGNC gene symbol. More than 28,000 human gene symbols and names have been assigned so far, including almost all of the protein coding genes. NCBI creates what might be thought of as interim codes. Older systems might send HGNC gene IDs without the "HGNC:" prefix. This is not following the guidance of the creators of the code system, and introduces ambiguity. So caution must be taken to confirm alignment. HGNC also provides an index on gene families/groups. GeneGroup IDs do not begin with "HGNC:", so care must be made to ensure alignment of concepts when viewing an HGNC ID from an older system that may be referring to the GeneID and not a gene group. For example, 588 refers to the HLA gene family, but HGNC:588 identifies the ATG12 gene. |
| web | www.ebi.ac.uk | HLA Nomenclature |
| web | hla.alleles.org | Human leukocyte antigen (HLA) is a gene family found in the Major Histocompatibility Complex (MHC) of Chromosome 6 in humans. This family includes more than 50 genes. A subset of these are commonly used as markers for histocompatibility testing for stem cell and solid organ transplantation, drug sensitivity, and disease association. The WHO Nomenclature Committee for Factors of the HLA System is responsible for a common nomenclature of HLA alleles, allele sequences, and quality control, to communicate histocompatibility typing information to match donors and recipients. An HLA allele is defined as any set of variations found on a sequence of DNA comprising a HLA gene. So, if there are five variations found in this one gene sequence, this set is defined as one allele (vs. the definition of an allele being the variation found between the test specimen and the reference along a contiguous stretch of DNA). In the case of HLA, the contiguous stretch of DNA represents the entire gene, and the variations do not need to be contiguous within the gene sequence. Each HLA allele name has a unique name consisting of the gene name followed by up to four fields, each containing at least two digits, separated by colons. There are also optional suffixes added to indicate expression status. Individual HLA genes may have thousands of different alleles. For the full specification, please go to this website: http://hla.alleles.org/nomenclature/naming.html. HLA nomenclature can also be used to represent sets of alleles that share sequence identity in the Antigen Recognition Site (ARS). G-groups are alleles that have identical DNA sequences in the ARS, while P-groups are alleles that have identical protein sequences in the ARS. These are described, respectively, in http://hla.alleles.org/alleles/g_groups.html and http://hla.alleles.org/alleles/p_groups.html . |
| web | hla.alleles.org | Human leukocyte antigen (HLA) is a gene family found in the Major Histocompatibility Complex (MHC) of Chromosome 6 in humans. This family includes more than 50 genes. A subset of these are commonly used as markers for histocompatibility testing for stem cell and solid organ transplantation, drug sensitivity, and disease association. The WHO Nomenclature Committee for Factors of the HLA System is responsible for a common nomenclature of HLA alleles, allele sequences, and quality control, to communicate histocompatibility typing information to match donors and recipients. An HLA allele is defined as any set of variations found on a sequence of DNA comprising a HLA gene. So, if there are five variations found in this one gene sequence, this set is defined as one allele (vs. the definition of an allele being the variation found between the test specimen and the reference along a contiguous stretch of DNA). In the case of HLA, the contiguous stretch of DNA represents the entire gene, and the variations do not need to be contiguous within the gene sequence. Each HLA allele name has a unique name consisting of the gene name followed by up to four fields, each containing at least two digits, separated by colons. There are also optional suffixes added to indicate expression status. Individual HLA genes may have thousands of different alleles. For the full specification, please go to this website: http://hla.alleles.org/nomenclature/naming.html. HLA nomenclature can also be used to represent sets of alleles that share sequence identity in the Antigen Recognition Site (ARS). G-groups are alleles that have identical DNA sequences in the ARS, while P-groups are alleles that have identical protein sequences in the ARS. These are described, respectively, in http://hla.alleles.org/alleles/g_groups.html and http://hla.alleles.org/alleles/p_groups.html . |
| web | www.pharmvar.org | PHARMVAR |
| web | bioinformatics.bethematchclinical.org | These principles were implemented in a technical specification by extending an existing XML based format for exchanging histocompatibility and immunogenetic genotyping data called Histoimmunogenetics Markup Language (HML) to include results from NGS methodologies. The resulting schema may be found here . The National Marrow Donor Program (NMDP)/Be The Match uses this format for reporting HLA genotyping from potential donors and for patients needing stem cell transplants. |
| web | schemas.nmdp.org | These principles were implemented in a technical specification by extending an existing XML based format for exchanging histocompatibility and immunogenetic genotyping data called Histoimmunogenetics Markup Language (HML) to include results from NGS methodologies. The resulting schema may be found here . The National Marrow Donor Program (NMDP)/Be The Match uses this format for reporting HLA genotyping from potential donors and for patients needing stem cell transplants. |
| web | www.ebi.ac.uk | Once HLA is typed, perhaps using targeted probes, exon sequencing, or full gene sequencing, the results are analyzed and assigned to one or more of the IMGT/HLA gene alleles using the HLA nomenclature . While the actual results may be a haplotype of exons, the results are assigned to an allele from a known list. This allows results from different HLA genotyping methodologies to be compared, for example for donor/recipient transplant matching. However, reducing the results to HLA nomenclature may introduce ambiguity in the actual results. For example, an allele may be reported at only a two-field protein level (e.g., HLA-B 57:01) which represents any of over forty DNA variants, while the actual data may represent a single full gene (e.g., HLA-B 57:01:01:05). The nomenclature may also over represent the quality of the data. For example, when a full gene allele name is assigned to the results, it is unknown how the data was captured (probes, exon-only sequence, or full gene sequencing). Methodology is captured separately in the report. Also, this system cannot easily capture novel alleles as it represents a closed-world of possible alleles. Earlier scientific articles and data may reference HLA serotypes (e.g., HLA-B57), recognizable by the absence of an asterisk in the name, which does not capture any molecular information. The IMGT/HLA allele database is updated every three months to account for the discovery of new alleles, and the deletion or renaming of old alleles. This is why it is important to include the IMGT/HLA version when the HLA allele is assigned. |
| web | hla.alleles.org | Once HLA is typed, perhaps using targeted probes, exon sequencing, or full gene sequencing, the results are analyzed and assigned to one or more of the IMGT/HLA gene alleles using the HLA nomenclature . While the actual results may be a haplotype of exons, the results are assigned to an allele from a known list. This allows results from different HLA genotyping methodologies to be compared, for example for donor/recipient transplant matching. However, reducing the results to HLA nomenclature may introduce ambiguity in the actual results. For example, an allele may be reported at only a two-field protein level (e.g., HLA-B 57:01) which represents any of over forty DNA variants, while the actual data may represent a single full gene (e.g., HLA-B 57:01:01:05). The nomenclature may also over represent the quality of the data. For example, when a full gene allele name is assigned to the results, it is unknown how the data was captured (probes, exon-only sequence, or full gene sequencing). Methodology is captured separately in the report. Also, this system cannot easily capture novel alleles as it represents a closed-world of possible alleles. Earlier scientific articles and data may reference HLA serotypes (e.g., HLA-B57), recognizable by the absence of an asterisk in the name, which does not capture any molecular information. The IMGT/HLA allele database is updated every three months to account for the discovery of new alleles, and the deletion or renaming of old alleles. This is why it is important to include the IMGT/HLA version when the HLA allele is assigned. |
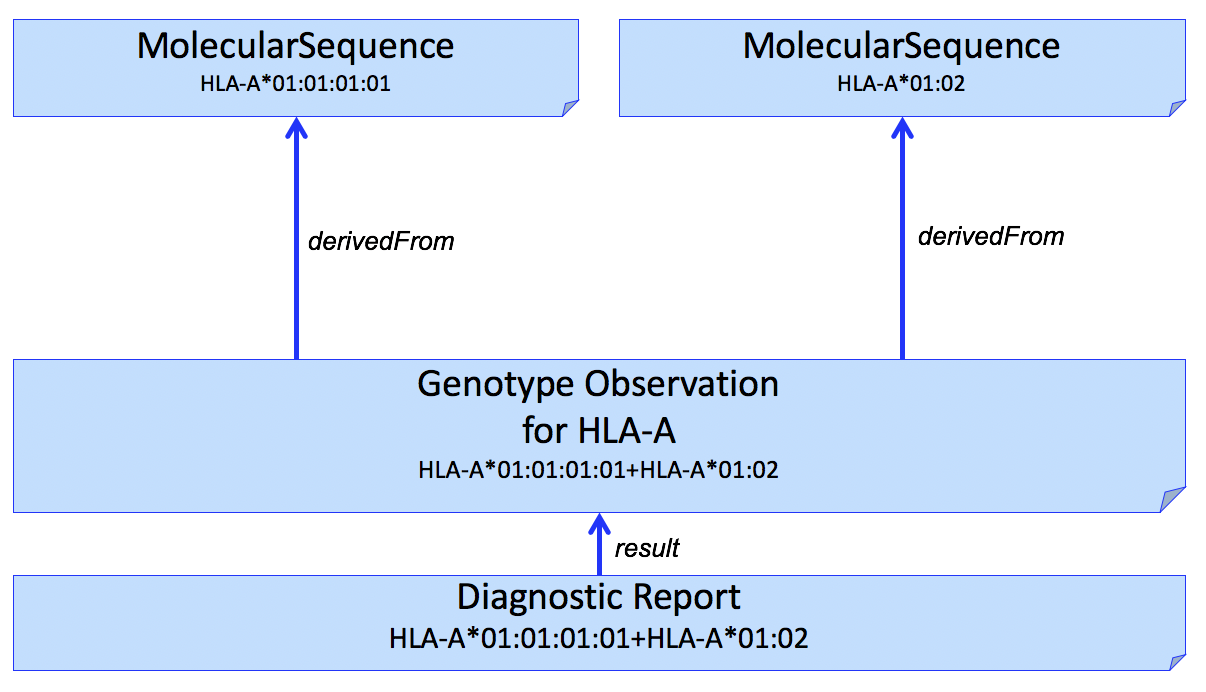
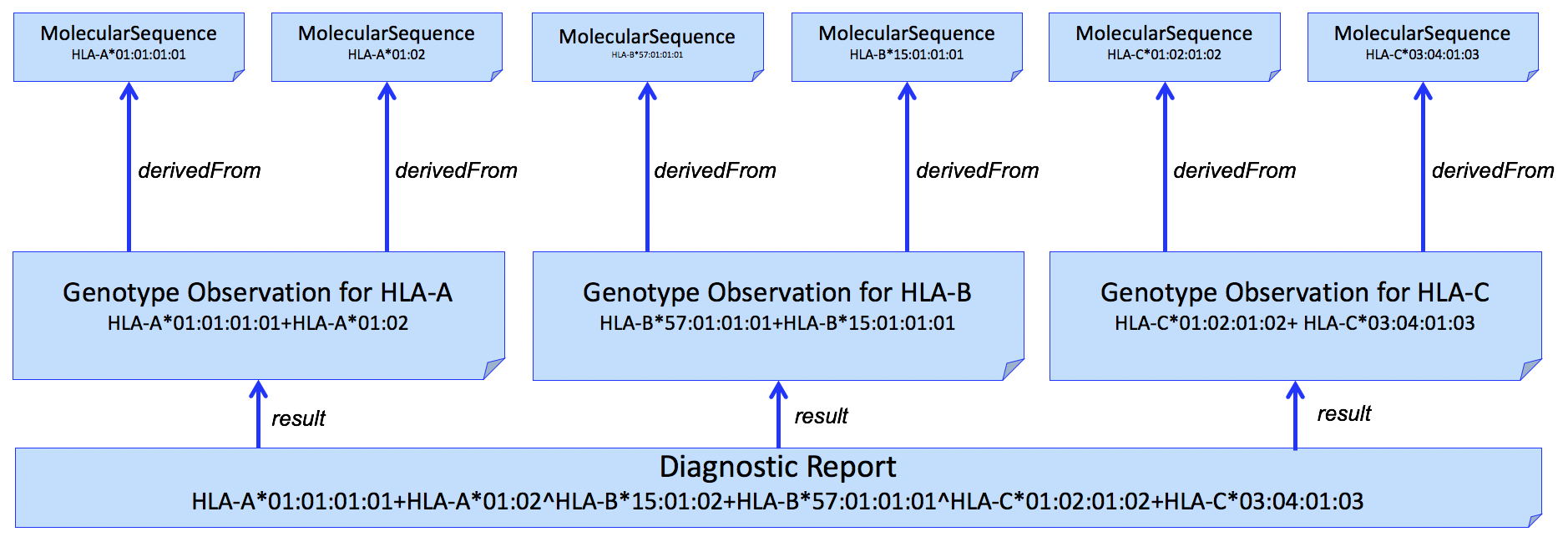
| web | www.ebi.ac.uk | In a simple case, the Genomic Report (DiagnosticReport) refers to a Genotype Observation for each HLA gene. The HLA alleles found in the genotype are typically assigned to alleles found in the Immuno Polymorphism Database (IPD) through the international ImMunoGeneTics (IMGT) project (found here ) and preferably expressed as a Genotype List String (GLString) using a GLString Code . |
| web | glstring.org | In a simple case, the Genomic Report (DiagnosticReport) refers to a Genotype Observation for each HLA gene. The HLA alleles found in the genotype are typically assigned to alleles found in the Immuno Polymorphism Database (IPD) through the international ImMunoGeneTics (IMGT) project (found here ) and preferably expressed as a Genotype List String (GLString) using a GLString Code . |
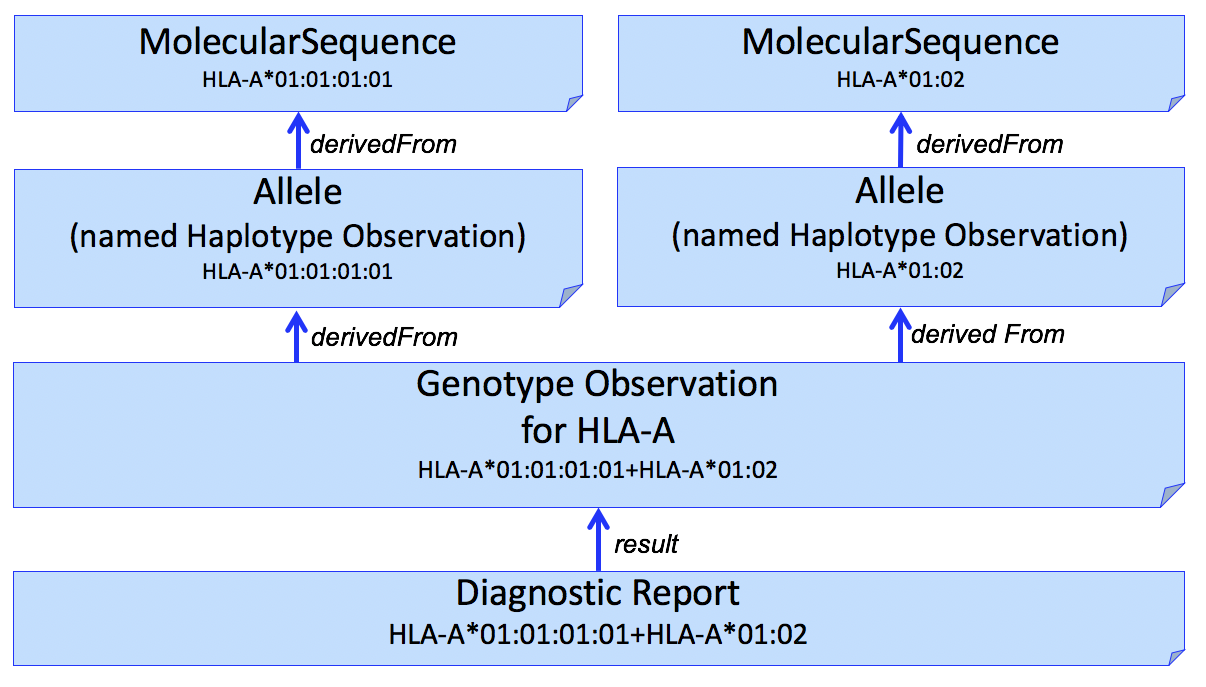
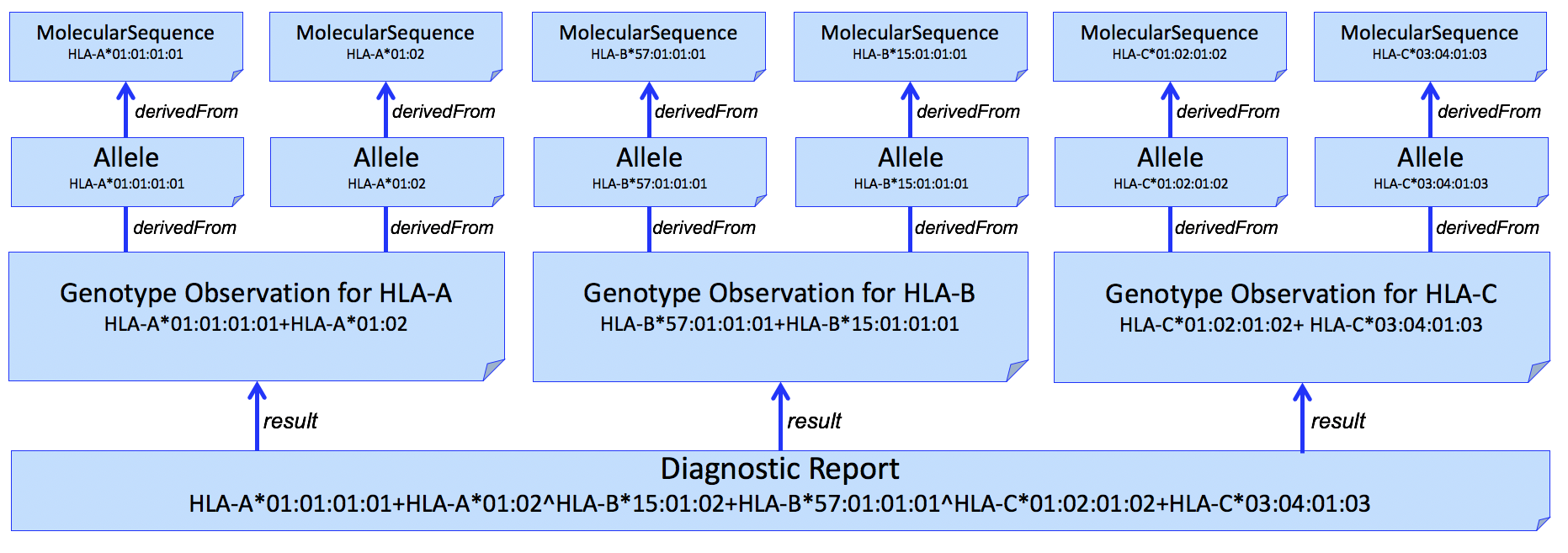
| web | docs.google.com | We use the Haplotype Observation for Allele assignment. In domains other than HLA (e.g., Pharmacogenomics), it is common to consider gene-level alleles as named haplotypes of a set of smaller variants ( Variation Modelling Collaboration Data Model and Specification ). This is not how the HLA community considers Haplotype, which is typically used to describe a set of gene-level alleles from different HLA genes that are found on the same DNA molecule. To be able to represent this latter description, this IG allows the representation "haplotypes of haplotypes." (example not shown) |
| web | bioinformatics.bethematchclinical.org | Allele ambiguity is often captured using NMDP Multiple Allele Codes . Because of limitations of this system, including introducing further ambiguity into the report, the GL String is preferred. This format uses a hierarchical set of operators to describe the relationships between alleles, lists of possible alleles, phased alleles, genotypes, lists of possible genotypes, and multilocus unphased genotypes, without losing typing information or increasing typing ambiguity. To use this format, the GL String Code system can be used, which embeds this GL String into a format containing the gene system, and version of the nomenclature used within the GL String. |
| web | glstring.org | Allele ambiguity is often captured using NMDP Multiple Allele Codes . Because of limitations of this system, including introducing further ambiguity into the report, the GL String is preferred. This format uses a hierarchical set of operators to describe the relationships between alleles, lists of possible alleles, phased alleles, genotypes, lists of possible genotypes, and multilocus unphased genotypes, without losing typing information or increasing typing ambiguity. To use this format, the GL String Code system can be used, which embeds this GL String into a format containing the gene system, and version of the nomenclature used within the GL String. |
| web | creativecommons.org |
Copyrights permitted under terms specified by Creative Commons Attribution 4.0 International (CC BY 4.0)
.Consistent with the terms of CC BY 4.0, HL7 is permitted to share, copy and redistribute the material in any medium or format and adapt, remix, transform, and build upon the material for its purposes as long as the CC BY 4.0 license terms are upheld. HL7 must must give appropriate credit, provide a link to the license, and indicate if changes were made. HL7 may do so in any reasonable manner, but not in any way that suggests the licensor endorses HL7 or it's use. And HL7 may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.
Show Usage
|
| web | omim.org |
Information that is created by or for the US government are within the public domain. Public domain information on the National Library of Medicine (NLM) Web pages may be freely distributed and copied. However, it is requested that in any subsequent use of this work, NLM be given appropriate acknowledgment.NOTE: This site contains resources which incorporate material contributed or licensed by individuals, companies, or organizations that may be protected by U.S. and foreign copyright laws. These include, but are not limited to PubMed Central (PMC) (see PMC Copyright Notice via https://www.ncbi.nlm.nih.gov/pmc/about/copyright/)
, Bookshelf (see Bookshelf Copyright Notice via https://www.ncbi.nlm.nih.gov/books/about/copyright/)
, OMIM (see OMIM Copyright Status via https://omim.org/help/copyright)
, and PubChem. All persons reproducing, redistributing, or making commercial use of this information are expected to adhere to the terms and conditions asserted by the copyright holder. Transmission or reproduction of protected items beyond that allowed by fair use ( https://www.copyright.gov/fls/fl102.html)(PDF)
as defined in the copyright laws requires the written permission of the copyright owners.For information on NCBI's policies and disclaimers for use, see here https://www.ncbi.nlm.nih.gov/home/about/policies/
.
Show Usage
|
| web | omim.org |
NCBI's Disclaimer and Copyright notice must be evident to users of your service.Information that is created by or for the US government on this site is within the public domain. Public domain information on the National Library of Medicine (NLM) Web pages may be freely distributed and copied. However, it is requested that in any subsequent use of this work, NLM be given appropriate acknowledgment.NOTE: This site contains resources which incorporate material contributed or licensed by individuals, companies, or organizations that may be protected by U.S. and foreign copyright laws. These include, but are not limited to PubMed Central (PMC) (see PMC Copyright Notice at https://www.ncbi.nlm.nih.gov/pmc/about/copyright/)
, Bookshelf (see Bookshelf Copyright Notice at https://www.ncbi.nlm.nih.gov/books/about/copyright/)
, OMIM (see OMIM Copyright Status at https://omim.org/help/copyright)
, and PubChem. All persons reproducing, redistributing, or making commercial use of this information are expected to adhere to the terms and conditions asserted by the copyright holder. Transmission or reproduction of protected items beyond that allowed by fair use ( https://www.copyright.gov/fls/fl102.html)(PDF)
as defined in the copyright laws requires the written permission of the copyright owners.General copyright, as stated on NCBI's GTR site ( https://www.ncbi.nlm.nih.gov/gtr/)
, is as included in the Copyright Statement.
Show Usage
|
| web | hpo.jax.org | That the Human Phenotype Ontology Consortium is acknowledged and cited properly. |
| web | github.com | That neither the content of the HPO file(s) nor the logical relationships embedded within the HPO file(s) be altered in any way. (Content additions and modifications have to be suggested using our issue tracker .) |
| web | www.human-phenotype-ontology.org | Users of the HPO should add the following statement to their online presence. This service/product uses the Human Phenotype Ontology (version information). Find out more at http://www.human-phenotype-ontology.org . We request that the HPO logo be included as well. |
| web | creativecommons.org |
The PharmVar database content is licensed under a Creative Commons Attribution-ShareAlike 4.0 International license
that allows for the sharing and adaptation of our information with proper attribution.See
https://www.pharmvar.org/terms-and-conditions
for Terms and Conditions.
Show Usage
|
| web | www.pharmvar.org |
The PharmVar database content is licensed under a Creative Commons Attribution-ShareAlike 4.0 International license
that allows for the sharing and adaptation of our information with proper attribution.See
https://www.pharmvar.org/terms-and-conditions
for Terms and Conditions.
Show Usage
|
| web | www.pharmvar.org | https://www.pharmvar.org/terms-and-conditions |
| web | genomebiology.com |
The Sequence Ontology: A tool for the unification of genome annotations. Eilbeck K., Lewis S.E., Mungall C.J., Yandell M., Stein L., Durbin R., Ashburner M. Genome Biology (2005) 6:R44
Please also include the version of SO used.Sequence Ontology data and data products are licensed under the Creative Commons Attribution 4.0 Unported License
.
Show Usage
|
| web | creativecommons.org |
The Sequence Ontology: A tool for the unification of genome annotations. Eilbeck K., Lewis S.E., Mungall C.J., Yandell M., Stein L., Durbin R., Ashburner M. Genome Biology (2005) 6:R44
Please also include the version of SO used.Sequence Ontology data and data products are licensed under the Creative Commons Attribution 4.0 Unported License
.
Show Usage
|
| web | www.karger.com |
The content on the Website including but not limited to the content of ISCN 2020 in whole or in parts, the title, the logo, graphic designs or adverts are the intellectual property of S. Karger AG (Basel) or published with permission from the legal copyright owner. All rights are reserved. Unless otherwise noted, no part of the content on the Website may be translated into other languages, reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, microcopying, or by any information storage and retrieval system, without permission in writing from Karger.For further information or permission requests please see the explanations under Rights and Permissions ( https://www.karger.com/Services/RightsPermissions)
.
Show Usage
|
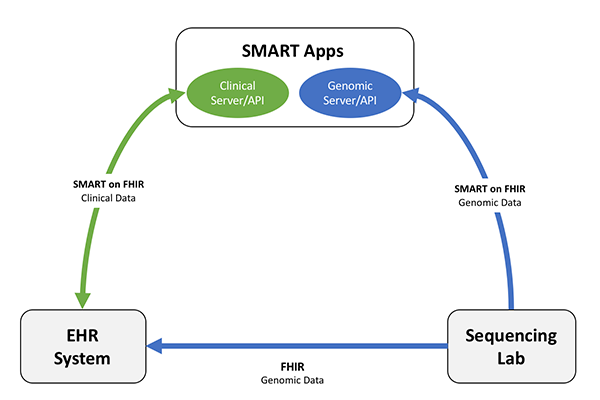
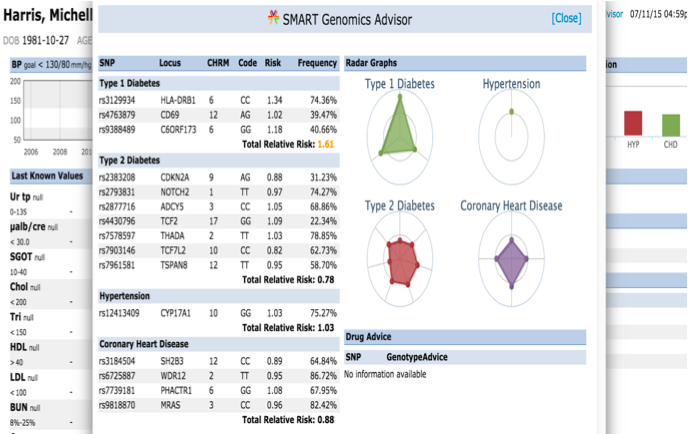
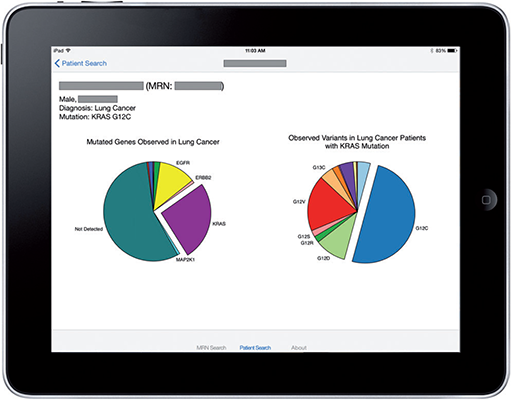
| web | smarthealthit.org | The September 2014 Informative Ballot ("HL7 Clinical Genomics, Domain Analysis Model: Clinical Sequencing Release 1") provided guiding use cases, which initially informed development of the initial Standard Genetics profile that is found in FHIR DSTU2. The same use cases also led to a second Project to develop a MolecularSequence resource ("Develop FHIR sequence resource for Clinical Genomics"). A preliminary effort to address these issues has been explored and published in context of the Substitutable Medical Applications and Reusable Technologies Platforms Project and described in an article (" SMART on FHIR Genomics: Facilitating standardized clinico-genomic apps "). |
| web | jamia.oxfordjournals.org | The September 2014 Informative Ballot ("HL7 Clinical Genomics, Domain Analysis Model: Clinical Sequencing Release 1") provided guiding use cases, which initially informed development of the initial Standard Genetics profile that is found in FHIR DSTU2. The same use cases also led to a second Project to develop a MolecularSequence resource ("Develop FHIR sequence resource for Clinical Genomics"). A preliminary effort to address these issues has been explored and published in context of the Substitutable Medical Applications and Reusable Technologies Platforms Project and described in an article (" SMART on FHIR Genomics: Facilitating standardized clinico-genomic apps "). |
| web | samtools.github.io | FHIR Genomics operations are based on the premise that genomic data, in FHIR format and/or some other format (e.g. VCF format ), are stored in a repository, either in or alongside an EHR, possibly along with phenotype annotations. The FHIR Genomics operations essentially 'wrap' the repository, presenting a uniform interface to applications, regardless of internal repository data structures. |
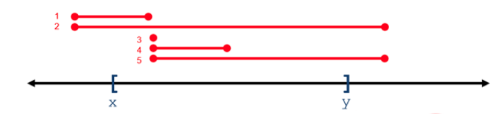
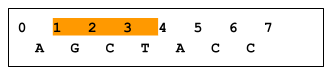
| web | www.biostars.org | For all operations, positional coordinates are zero-based . Some operations return variants that overlap a 0-based interval range of integers, whereas some operations return variants that subsume a range. In the following figure, variants 1-5 overlap or intersect with the X..Y range, whereas only variant 2 subsumes the X..Y range. |
| web | github.com | Several groups have identified edge cases that pose genome assembly conversion challenges (e.g. see PharmGKB's PharmCAT posting ; Biostars posting ). For example, NC_000001.11:145923295:C:C (build 38 representation) does not convert to a corresponding build 37 representation using NCBI Variation Services . As a result, there is no requirement that servers normalize all variants against a single build. |
| web | www.biostars.org | Several groups have identified edge cases that pose genome assembly conversion challenges (e.g. see PharmGKB's PharmCAT posting ; Biostars posting ). For example, NC_000001.11:145923295:C:C (build 38 representation) does not convert to a corresponding build 37 representation using NCBI Variation Services . As a result, there is no requirement that servers normalize all variants against a single build. |
| web | genviz.org | Many efficient and open source liftover tools exist (e.g. many are listed here ). As with variant liftover, translating a region between builds can also fail. For example, attempting to liftover NC_000001.11:145923295-145923296 (build 38 range) into a build 37 range with the UCSD LiftOver tool fails, because the region is partially deleted in build 37. In the (very uncommon) case of a failed lift over, a server can widen the query region as necessary in order to have a successful liftover. For example, the widened build 38 range NC_000001.11:145923285-145923306 will successfully translate into the build 37 range NC_000001.10:145511787-145511807. In the rare case where a server is storing variants aligned to multiple builds and the server is unable to liftover the query region, the server can return a response code of 422 "ERROR: Failed LiftOver", in which case the onus falls to the client to modify the query region. |
| web | genome.ucsc.edu | Many efficient and open source liftover tools exist (e.g. many are listed here ). As with variant liftover, translating a region between builds can also fail. For example, attempting to liftover NC_000001.11:145923295-145923296 (build 38 range) into a build 37 range with the UCSD LiftOver tool fails, because the region is partially deleted in build 37. In the (very uncommon) case of a failed lift over, a server can widen the query region as necessary in order to have a successful liftover. For example, the widened build 38 range NC_000001.11:145923285-145923306 will successfully translate into the build 37 range NC_000001.10:145511787-145511807. In the rare case where a server is storing variants aligned to multiple builds and the server is unable to liftover the query region, the server can return a response code of 422 "ERROR: Failed LiftOver", in which case the onus falls to the client to modify the query region. |
| web | github.com | An open source reference implementation of the operations is provided here . |
| web | emerge-fhir-spec.readthedocs.io | Notable changes from the eMERGE III FHIR Template docs include: |
| web | www.biorxiv.org | For further information about the eMERGE Program, see: Pre-Print Journal Article: "Genomic Considerations for FHIR; eMERGE Implementation Lessons" and eMERGE Network Website . |
| web | emerge-network.org | For further information about the eMERGE Program, see: Pre-Print Journal Article: "Genomic Considerations for FHIR; eMERGE Implementation Lessons" and eMERGE Network Website . |
| web | varnomen.hgvs.org | Proper usage of HGVS contains the reference sequence identifier followed by ':g.' for genomic or ':c.' for a coding sequence. In HGVS notation, the "=" (equals) is used to indicate a sequence was tested but found unchanged [ref] . |
|
4-pharmacogenomics-fig1-implications.png
|
|
Report_Page1.svg
|
|
Report_Page2.svg
|
|
Report_Page3.svg
|
|
Report_Page4.svg
|
|
Somatic_General_Guidance.svg
|
|
Somatic_Report_Overview.svg
|
|
cgapps-StructureSMARTApps-fg1.png
|
|
cgapps-genomicsAdviser-fg2.png
|
|
cgapps-precisionCancerMedicine-fg3.png
|
|
cgapps-precisionCancerMedicine-fg4.png
|
|
cgapps-precisionCancerMedicine-fg5.png
|
|
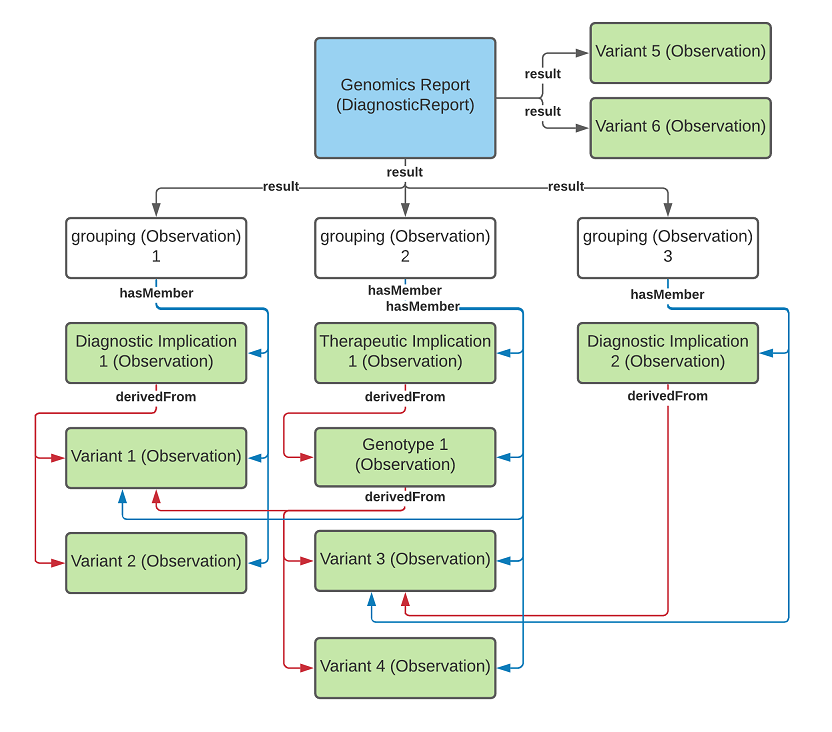
diagnostic-report-MixedUseOfGrouper.png
|
|
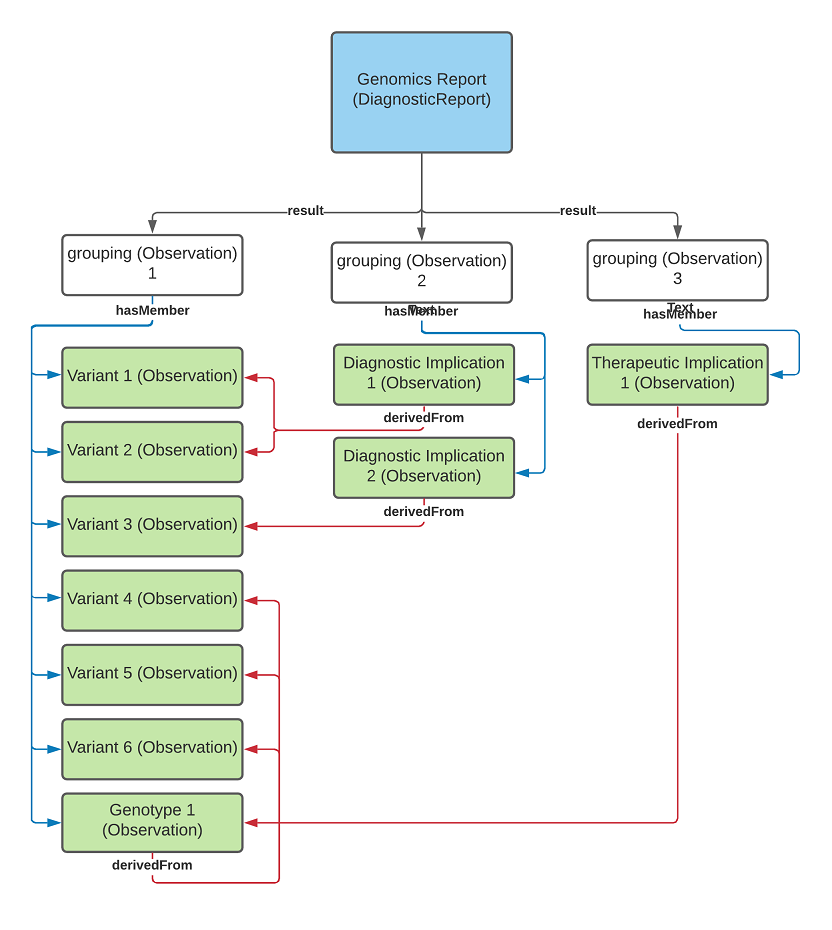
diagnostic-report-groupingObservations.png
|
|
diagnostic-report-no-grouping.png
|
|
find-variants-fig1.png
|
|
general-genomic-findings.png
|
|
general-genomic-implications.png
|
|
general-observations.png
|
|
general-relationships.png
|
|
general-report-overview.png
|
|
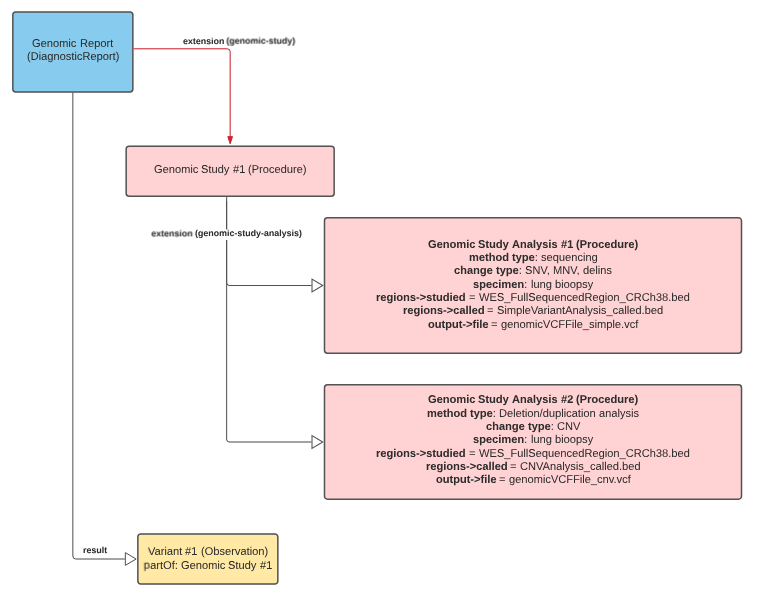
genomicStudy.png
|
|
histocompatibility-fig1-hla-a-genotype.png
|
|
histocompatibility-fig2-hla-a-genotype-allele.png
|
|
histocompatibility-fig3-hla-abc-genotype.png
|
|
histocompatibility-fig4-hla-abc-genotype-allele.png
|
|
implication-and-or-ex1.jpg
|
|
implication-and-or-ex2.jpg
|
|
implication-and-or-ex3.jpg
|
|
implication-and-or-ex4.jpg
|
|
implications.png
|
|
molecularConsequences2.png
|
|
molecularConsequences3.png
|
|
pgx-guidance-figure-1.jpg
|
|
pgx-guidance-figure-2.jpg
|
|
pgx-guidance-figure-3.jpg
|
|
tree-filter.png
|
|
zero-based.png
|