
Genomics Reporting Implementation Guide
4.0.0-cibuild - CI Build
![]()
Genomics Reporting Implementation Guide
4.0.0-cibuild - CI Build
![]()
Genomics Reporting Implementation Guide, published by HL7 International / Clinical Genomics. This guide is not an authorized publication; it is the continuous build for version 4.0.0-cibuild built by the FHIR (HL7® FHIR® Standard) CI Build. This version is based on the current content of https://github.com/HL7/genomics-reporting/ and changes regularly. See the Directory of published versions
| Page standards status: Informative |
FHIR Operations are a supplement to standard queries, and are used to fulfill a number of purposes, such as where the server needs to play an active role in formulating the content of the response, where the intended purpose is to cause side effects such as the creation of new resources, and for data normalization to abstract away from variability in variant representation. This is particularly relevant in genomics, where a tremendous amount of raw data exists in non-FHIR formats, and where variability in variant representation is a known barrier to matching a patient's genetic profile against knowledge bases – for clinical trials matching, for precision medication selection, etc.
FHIR Genomics operations are based on the premise that genomic data, in FHIR format and/or some other format (e.g. VCF format), are stored in a repository, either in or alongside an EHR, possibly along with phenotype annotations. The FHIR Genomics operations essentially 'wrap' the repository, presenting a uniform interface to applications, regardless of internal repository data structures.
A common use case driving the operations is the notion of an application (e.g. a SMART-ON-FHIR clinical genomics App, a clinical decision support application, an EHR screen) needing specific genotype or phenotype information, for a patient or a population. Applications have diverse needs, such as matching a cancer patient to available clinical trials based on identified somatic variants; screening for actionable hereditary conditions; identifying a risk for adverse medication reactions based on pharmacogenomic variants; updating a patient's risk as knowledge of their variants evolves; and more. A goal for FHIR Genomics operations is to ultimately support any and all of these clinical scenarios.
In scope are clinical genomics operations. In the future, operations supporting genomics research, variant calling and annotation, and knowledge base lookups may be added. We further categorize clinical genomics operations along two orthogonal axes - subject vs. population, and genotype vs. phenotype. For example, the 'find-subject-variants' operation retrieves genotype information for a single subject; whereas the 'find-population-tx-implications' retrieves a count or list of patients having specific phenotypes (such as being intermediate metabolizers of clopidogrel).
| Subject Operations | Population Operations | |
|---|---|---|
| Genotype Operations | ||
| simple variants | find-subject-variants; find-subject-specific-variants | find-population-specific-variants |
| structural variants | find-subject-structural-intersecting-variants; find-subject-structural-subsuming-variants | find-population-structural-intersecting-variants; find-population-structural-subsuming-variants |
| haplotype/genotypes | find-subject-haplotypes; find-subject-specific-haplotypes | find-population-specific-haplotypes |
| Phenotype Operations | ||
| therapeutic implications | find-subject-tx-implications | find-population-tx-implications |
| diagnostic implications | find-subject-dx-implications | find-population-dx-implications |
| molecular consequences | find-subject-molecular-consequences | find-population-molecular-consequences |
| Metadata Operations | ||
| study metadata | find-study-metadata |
| Operation | Description |
|---|---|
| find-subject-variants | Determine if simple variants are present that overlap range(s). |
| find-subject-specific-variants | Determine if specified simple variants are present. |
| find-subject-structural-intersecting-variants | Determine if structural variants are present that overlap range(s). |
| find-subject-structural-subsuming-variants | Determine if structural variants are present that fully subsume a range. |
| find-subject-haplotypes | Retrieve haplotypes/genotypes for specified genes. |
| find-subject-specific-haplotypes | See if specified haplotypes/genotypes are present. |
| find-subject-tx-implications | Retrieves genetic therapeutic implications for variants/haplotypes/genotypes. |
| find-subject-dx-implications | Retrieves genetic diagnostic implications for variants. |
| find-subject-molecular-consequences | Retrieves molecular consequences of a DNA variant. |
| find-population-specific-variants | Retrieve count or list of patients having specified variants. |
| find-population-structural-intersecting-variants | Retrieve count or list of patients having structural intersecting variants in specified regions. |
| find-population-structural-subsuming-variants | Retrieve count or list of patients having structural subsuming variants in specified regions. |
| find-population-specific-haplotypes | Retrieve count or list of patients having specified genotypes/haplotypes. |
| find-population-tx-implications | Retrieve count or list of patients having therapeutic implications. |
| find-population-molecular-consequences | Retrieve count or list of patients having molecular consequences. |
| find-population-dx-implications | Retrieve count or list of patients having diagnostic implications. |
| find-study-metadata | Retrieve metadata about sequencing studies performed on a subject. |
Genomics is a complex and highly dynamic field. We provide general guidance applicable to all FHIR Genomics operations here.
| Many operations have coded elements as input parameters. In such cases, the description of the parameter will include 'must be in token or codesystem | code format'. A token is a string that should be used by the server to perform case-insensitive search through text, display, code, and/or codeSystem. Servers may optionally broaden the use of a token to also perform close to exact searching. If supplied parameter is in codeSystem | code format, server should only perform case-sensitive code- and codeSystem-specific retrieval. |
Genomic data repositories will often contain the results of more than one test on a given patient. A patient with cancer may be sequenced repeatedly over time, e.g. at initial diagnosis and again if the tumor recurs; a patient with a diagnostic odyssey may have one test focused on the identification of simple variants and another test focused on structural variants; a patient with multiple metastases may have multiple tumor specimens that are sequenced; a patient may have both a germline sequencing test and a tumor sequencing test; etc.
In such cases, it is important that FHIR Genomics operations be able to specify query filters, so that only the clinically relevant subset of results are returned. Currently, operations support four filters, all of which are optional, and all of which can be determined using the find-study-metadata operation:
FHIR Search defines general AND/OR logic in FHIR queries: A logical 'AND' is achieved by repeating a query parameter (e.g. '/Patient?language=FR&language=NL'), whereas a logical 'OR' is achieved via a comma-separated list of values for a single query parameter (e.g. '/Patient?language=FR,NL'). Unless stated otherwise, all FHIR Genomics Operations support 'OR' logic, and only support 'AND' logic for date fields (e.g. '…&testDateRange=ge2020-06-01&testDateRange=le2020-08-01'). Parameters that have a cardinality greater than one can be populated with a comma-separated list of values that represent a logical 'OR'.
While sources vary in how they differentiate simple vs. structural variants, here we differentiate based on endpoint precision. Simple variant operations (find-subject-variants, find-subject-specific-variants) assume precise endpoints and return variants in a 'VCF-like' format, with 'exact-start-end' coordinates. Structural variant operations (find-subject-structural-intersecting-variants, find-subject-structural-subsuming-variants) assume imprecise endpoints and return variants per structural variant reporting guidelines with 'outer-start-end' and 'inner-start-end' coordinates.
As our understanding of genomics grows, we continue to recognize the clinical significance of variations not only within protein-coding gene exons, but also in introns and intergenic regions. To enable a consistent approach to query, whether for variants within or outside a gene, these operations are generally based on regions as parameters, and not gene names or identifiers. As a result, to query for variants within a specific gene, an application must determine the position of that gene, and supply the position as a parameter. For instance, assume an App wants to use the 'find-subject-variants' operation to see if a subject has any variants in the APC gene. On human genome reference assembly GRCh38, the APC gene is located on chromosome 5 (reference sequence NC_000005.10), at position 112707497-112846239. Thus, the query would be for variants that overlap NC_000005.10:112707497-112846239. The definitive location of genes based on human genome resources at the NCBI can be found here.
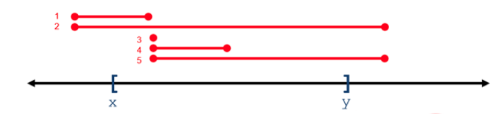
For all operations, positional coordinates are zero-based. Some operations return variants that overlap a 0-based interval range of integers, whereas some operations return variants that subsume a range. In the following figure, variants 1-5 overlap or intersect with the X..Y range, whereas only variant 2 subsumes the X..Y range.
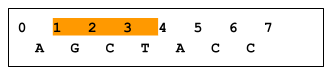
A zero-based coordinate system has an inclusive start and an exclusive end. For example, in the following figure, bases GCT are in range 1..4, whereas base A is not in range 1..4. As a result, a variant at position X is considered to intersect the X..Y range, whereas a variant at position Y does not. (Note that an insertion that starts before X, regardless of length, would not overlap X..Y, and would therefore not be returned). Imprecise implementations are allowed, where results contain some records outside a requested range.
Optionality in the Variant profile allows for different implementations to represent variants in different ways. For instance, the following variant representations are synonymous:
Implicit in these FHIR Genomics operations is that variants in requested regions are returned regardless of how they are formatted/represented/stored in a server. While the specific implementation of an Operation is outside the scope of HL7, we do provide guidance on how the above representations might be normalized such that any would be found and returned in a request for variants overlapping, say, the region of the LDLR gene (NC_000019.10:11089362-11133830). There are likely other effective normalization strategies beyond what is described here.
One approach to normalization is to convert all representations to a canonical form, such as the NCBI's Sequence Position Deletion Insertion (SPDI) format. Variant queries then only need to query a single format.
'SPDI' is the NCBI's variation notation for variants with known breakpoints. The notation represents an observed variant sequence using deleted and inserted sequences at a given position in a reference sequence. SPDI notation uses four fields and is written out as four elements delimited by colons S:P:D:I, where S=SequenceId; P=Position, a 0-based coordinate for where the Deleted Sequence starts; D=DeletedSequence, and I=InsertedSequence. Variation Services only support variants where the coordinates of both the upstream and downstream breakpoints are known (e.g. single nucleotide change, deletions at precise coordinates). Such variants can be encoded precisely using the SPDI notation.
NCBI Variation Services provide a rich set of APIs that can be used to normalize variants from many formats (e.g. HGVS, VCF) into SPDI, and to normalize variants in SPDI into a canonical SPDI format. The variant above, in canonical SPDI format, resolves to this: NC_000019.10:11089559:G:A, where it can easily be determined that it overlaps the requested region (NC_000019.10:11,089,362-11,133,830).
LiftOver is a process whereby a genome position is converted from one genome assembly to another genome assembly. It is the process that, among other things, allows us to determine that these two variants are the same:
Several groups have identified edge cases that pose genome assembly conversion challenges (e.g. see PharmGKB's PharmCAT posting; Biostars posting). For example, NC_000001.11:145923295:C:C (build 38 representation) does not convert to a corresponding build 37 representation using NCBI Variation Services. As a result, there is no requirement that servers normalize all variants against a single build.
Rather, where a server is storing variants aligned to multiple builds (and hasn't normalized all variants against a single build), it will be necessary for the server to lift over the query region into corresponding regions in other builds. For example, a query for variants in NC_000001.11:145507556-145513536 (build 38 range) may also need to query for variants in NC_000001.10:145921556-145927537 (build 37 range) in order to gather variants expressed against build 38 and build 37, respectively.
Many efficient and open source liftover tools exist (e.g. many are listed here). As with variant liftover, translating a region between builds can also fail. For example, attempting to liftover NC_000001.11:145923295-145923296 (build 38 range) into a build 37 range with the UCSD LiftOver tool fails, because the region is partially deleted in build 37. In the (very uncommon) case of a failed lift over, a server can widen the query region as necessary in order to have a successful liftover. For example, the widened build 38 range NC_000001.11:145923285-145923306 will successfully translate into the build 37 range NC_000001.10:145511787-145511807. In the rare case where a server is storing variants aligned to multiple builds and the server is unable to liftover the query region, the server can return a response code of 422 "ERROR: Failed LiftOver", in which case the onus falls to the client to modify the query region.
An open source reference implementation of the operations is provided here.
IG © 2025+ HL7 International / Clinical Genomics. Package hl7.fhir.uv.genomics-reporting#4.0.0-cibuild based on FHIR 4.0.1. Generated 2026-01-30
Links: Table of Contents |
QA Report
| Version History |
 |
Propose a change
|
Propose a change