Note to Implementers:
Molecular Sequence resource will be replaced by the Molecular Definition resource. Current page is temporarily available for referencing and review purposes.
10.7.1 Scope and Usage
The MolecularSequence resource is designed for representing molecular sequences. It can represent the sequence in different ways, allowing implementations to adopt the most effective one for their use case.
It is strongly encouraged to provide as much information in this resource for any reported sequences, because receiving systems (e.g. discovery research, outcomes analysis, and public health reporting) may use this information to normalize sequences over time or across sources. However, these data should not be used to dynamically correct/change sequence representations for clinical use outside of the laboratory, due to insufficient information.
The MolecularSequence resource is designed to represent a single sequence in an instance. Each sequence might have multiple representations, but implementers SHALL ensure all representations are for the same sequence. This means that if a single MolecularSequence instance contains a literal, two file files, and a relative, all four of those representations must represent the same sequence. This can be a challenge across systems, as semantic equivalency of sequences cannot be guaranteed unless there is an agreed upon standard between sending and receiving systems.
10.7.2 Boundaries and Relationships
The MolecularSequence resource should only be used to capture a molecular sequence. It will not be used for other entities such as variant, variant annotations, genotypes, haplotypes, etc. Those concepts will be captured in Observation profiles found in the Genomics Reporting Implementation Guide . The sequence that was observed that led to the identification of those concepts can be delivered with this resource, and will be referenced by those observations.
MolecularSequence will not be used to capture data such as precise read of DNA sequences and sequence alignment are not included; such data may be accessible through references to GA4GH (Global Alliance for Genomics and Health) API, and may be referenced to by the file element.
The coordinate system used to define the edited intervals on the starting sequence. Coordinate systems are usually 0- or 1-based Binding: LL5323-2 (Extensible)
@prefix fhir: <http://hl7.org/fhir/> .
[ a fhir:MolecularSequence;
fhir:nodeRole fhir:treeRoot; # if this is the parser root
# from Resource: .id, .meta, .implicitRules, and .language
# from DomainResource: .text, .contained, .extension, and .modifierExtension
fhir:identifier ( [ Identifier ] ... ) ; # 0..* Unique ID for this particular sequence
fhir:type[ code ] ; # 0..1 aa | dna | rna
fhir:literal( [ # 0..* A literal representation of a Molecular Sequence
fhir:sequenceValue[ string ] ; # 1..1 The primary (linear) sequence, expressed as a literal string
] ... ) ;
fhir:file ( [ Attachment ] ... ) ; # 0..* Embedded file or a link (URL) which contains content to represent the sequence
fhir:relative( [ # 0..* A Molecular Sequence that is represented as an ordered series of edits on a specified starting sequence
fhir:startingSequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence that serves as the starting sequence, on which edits will be applied
fhir:edit( [ # 0..* An edit (change) made to a sequence
fhir:editOrder[ integer ] ; # 0..1 The order of this edit, relative to other edits on the starting sequence
fhir:coordinateSystem[ CodeableConcept ] ; # 1..1 The coordinate system used to define the edited intervals on the starting sequence. Coordinate systems are usually 0- or 1-based
fhir:start[ integer ] ; # 1..1 The start coordinate of the interval that will be edited
fhir:end[ integer ] ; # 1..1 The end coordinate of the interval that will be edited
fhir:replacementSequence[ Reference(MolecularSequence) ] ; # 1..1 The sequence that defines the replacement sequence used in the edit operation
fhir:replacedSequence[ Reference(MolecularSequence) ] ; # 0..1 The sequence on the 'starting' sequence for the edit operation, defined by the specified interval, that will be replaced during the edit
] ... ) ;
] ... ) ;
fhir:extracted( [ # 0..* A Molecular Sequence that is represented as an extracted portion of a different Molecular Sequence
fhir:startingSequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence that serves as the parent sequence, from which the intended sequence will be extracted
fhir:start[ integer ] ; # 1..1 The start coordinate (on the parent sequence) of the interval that defines the subsequence to be extracted
fhir:end[ integer ] ; # 1..1 The end coordinate (on the parent sequence) of the interval that defines the subsequence to be extracted
fhir:coordinateSystem[ CodeableConcept ] ; # 1..1 The coordinate system used to define the interval that defines the subsequence to be extracted. Coordinate systems are usually 0- or 1-based
fhir:reverseComplement[ boolean ] ; # 0..1 A flag that indicates whether the extracted sequence should be reverse complemented
] ... ) ;
fhir:repeated( [ # 0..* A Molecular Sequence that is represented as a repeated sequence motif
fhir:sequenceMotif[ Reference(MolecularSequence) ] ; # 1..1 The sequence that defines the repeated motif
fhir:copyCount[ integer ] ; # 1..1 The number of repeats (copies) of the sequence motif
] ... ) ;
fhir:concatenated[ # 0..1 A Molecular Sequence that is represented as an ordered concatenation of two or more Molecular Sequences
fhir:sequenceElement( [ # 1..* One element of a concatenated Molecular Sequence
fhir:sequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence corresponding to this element
fhir:ordinalIndex[ integer ] ; # 1..1 The ordinal position of this sequence element within the concatenated Molecular Sequence
] ... ) ;
] ;
]
The coordinate system used to define the edited intervals on the starting sequence. Coordinate systems are usually 0- or 1-based Binding: LL5323-2 (Extensible)
@prefix fhir: <http://hl7.org/fhir/> .
[ a fhir:MolecularSequence;
fhir:nodeRole fhir:treeRoot; # if this is the parser root
# from Resource: .id, .meta, .implicitRules, and .language
# from DomainResource: .text, .contained, .extension, and .modifierExtension
fhir:identifier ( [ Identifier ] ... ) ; # 0..* Unique ID for this particular sequence
fhir:type[ code ] ; # 0..1 aa | dna | rna
fhir:literal( [ # 0..* A literal representation of a Molecular Sequence
fhir:sequenceValue[ string ] ; # 1..1 The primary (linear) sequence, expressed as a literal string
] ... ) ;
fhir:file ( [ Attachment ] ... ) ; # 0..* Embedded file or a link (URL) which contains content to represent the sequence
fhir:relative( [ # 0..* A Molecular Sequence that is represented as an ordered series of edits on a specified starting sequence
fhir:startingSequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence that serves as the starting sequence, on which edits will be applied
fhir:edit( [ # 0..* An edit (change) made to a sequence
fhir:editOrder[ integer ] ; # 0..1 The order of this edit, relative to other edits on the starting sequence
fhir:coordinateSystem[ CodeableConcept ] ; # 1..1 The coordinate system used to define the edited intervals on the starting sequence. Coordinate systems are usually 0- or 1-based
fhir:start[ integer ] ; # 1..1 The start coordinate of the interval that will be edited
fhir:end[ integer ] ; # 1..1 The end coordinate of the interval that will be edited
fhir:replacementSequence[ Reference(MolecularSequence) ] ; # 1..1 The sequence that defines the replacement sequence used in the edit operation
fhir:replacedSequence[ Reference(MolecularSequence) ] ; # 0..1 The sequence on the 'starting' sequence for the edit operation, defined by the specified interval, that will be replaced during the edit
] ... ) ;
] ... ) ;
fhir:extracted( [ # 0..* A Molecular Sequence that is represented as an extracted portion of a different Molecular Sequence
fhir:startingSequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence that serves as the parent sequence, from which the intended sequence will be extracted
fhir:start[ integer ] ; # 1..1 The start coordinate (on the parent sequence) of the interval that defines the subsequence to be extracted
fhir:end[ integer ] ; # 1..1 The end coordinate (on the parent sequence) of the interval that defines the subsequence to be extracted
fhir:coordinateSystem[ CodeableConcept ] ; # 1..1 The coordinate system used to define the interval that defines the subsequence to be extracted. Coordinate systems are usually 0- or 1-based
fhir:reverseComplement[ boolean ] ; # 0..1 A flag that indicates whether the extracted sequence should be reverse complemented
] ... ) ;
fhir:repeated( [ # 0..* A Molecular Sequence that is represented as a repeated sequence motif
fhir:sequenceMotif[ Reference(MolecularSequence) ] ; # 1..1 The sequence that defines the repeated motif
fhir:copyCount[ integer ] ; # 1..1 The number of repeats (copies) of the sequence motif
] ... ) ;
fhir:concatenated[ # 0..1 A Molecular Sequence that is represented as an ordered concatenation of two or more Molecular Sequences
fhir:sequenceElement( [ # 1..* One element of a concatenated Molecular Sequence
fhir:sequence[ Reference(MolecularSequence) ] ; # 1..1 The Molecular Sequence corresponding to this element
fhir:ordinalIndex[ integer ] ; # 1..1 The ordinal position of this sequence element within the concatenated Molecular Sequence
] ... ) ;
] ;
]
This resource supports three patterns for representing a sequence of interest:
By providing a literal string of IUPAC codes representing nucleotides or amino acids.
By linking to a formatted file or link containing the sequence information (e.g. FASTA file or GA4GH sequence repository).
By providing a list of edits from a starting sequence.
The MolecularSequence resource is designed to represent a single sequence in an instance. Each sequence might have multiple representations, but implementers SHALL ensure all representations are for the same sequence.
10.7.5.1.1 Sequence as a literal string
literal: This string element can be used to hold the sequence as a string of characters.
10.7.5.1.2 Sequence as a file or URL
file: This Attachment is used to refer to the sequence as embedded file content or via a URL reference.
This method can be used to refer to sequence data from in an external source. If the sequence is referring to a GA4GH repository, the file.url should refer to a GA4GH compliant endpoint that conforms to GA4GH data models.
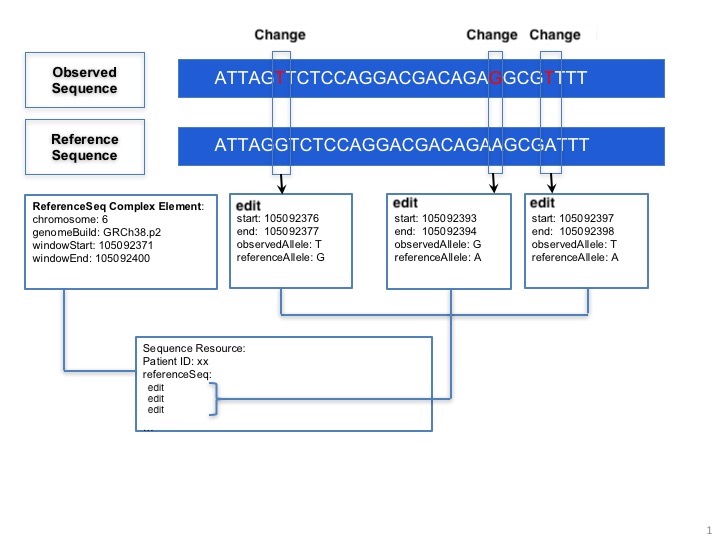
10.7.5.1.3 Sequence as a series of edits from a known sequence
relative: This complex element is used for encoding sequence. When the information of starting sequence and edits are provided, the observed sequence will be derived. Here is a picture below:
10.7.5.1.3.1 Composing multiple relative sequences into one new sequence
relative.ordinalPosition: Indicates the order in which the sequence should be considered when putting multiple relative instances together.
relative.sequenceRange: Indicates the nucleotide range in the composed sequence when multiple relative instances are used together.
These attributes help to clarify what sequence is being represented with less computation/inference on the recipient side. Implementers SHOULD use sequenceRange first to determine order as the most reliable. If sequenceRange is not present then ordinalPosition SHOULD be used. Finally, if both sequenceRange and ordinalPosition are absent, then the order of the relative data elements SHOULD be used to calculate a composition. It is the responsibility of the data sender to ensure the message can be consistently understood. Additionally, gaps in sequenceRange are considered intentional (i.e. the composed sequence contains a sequence of N's, the placeholder nucleotide, for the gap range).
10.7.5.1.3.2 Representing the Starting Sequence
relative.startingSequence: There are four optional ways to represent a starting sequence in MolecularSequence resource:
relative.startingSequence.sequenceCodeableConcept: Starting sequence id in public database;
relative.startingSequence.sequenceReference: Reference to starting sequence stored in another sequence entity;
relative.startingSequence.genomeAssembly, relative.startingSequence.chromosome: The combination of genome assembly and chromosome.
The relative.startingSequence.windowStart and relative.startingSequence.windowEnddefines a range from the starting sequence that is used to define a subsequence used as the starting sequence.
10.7.5.1.3.3 Coordinate System
When saving the sequence information, the nucleic acid will be numbered with order. Some representations use a 0-based system (e.g. GA4GH API, BAM files) while some use a 1-based system (e.g. VCF file format). The element coordinateSystem contains this information.
relative.coordinateSystem binds to a LOINC answer list, please review those answers here as well as the detailed description found here .
10.7.5.1.3.4 Choice of Strand
There are many considerations concerning the directionality of DNA or RNA. Here we are using relative.startingSequence.orientation and relative.startingSequence.strand. Orientation represents the sense of the sequence, which has different meanings depending on the type. Strand represents the sequence writing order. Watson strand refers to 5' to 3' top strand (5' -> 3'), whereas Crick strand refers to 5' to 3' bottom strand (3' <- 5').
Only two possible values can be made by strand, watson and crick. Since the directionality of the sequence string might be represented in different ways in different omics scenario, below are examples of how to map other expressions into its correlated value:
Watson
Crick
5′-to-3′ direction
3′-to-5′ direction
+1
-1
Sense
Antisense
Positive
Negative
10.7.5.2 Character usage for sequence as strings
There are attributes where the sequence is represented as a string of characters.

 Diagnostics
Diagnostics



 |
Propose a change
|
Propose a change 