
Adverse Event Clinical Research R4 Backport
1.0.1 - STU1
![]()
Adverse Event Clinical Research R4 Backport
1.0.1 - STU1
![]()
Adverse Event Clinical Research R4 Backport, published by HL7 International / Biomedical Research and Regulation. This guide is not an authorized publication; it is the continuous build for version 1.0.1 built by the FHIR (HL7® FHIR® Standard) CI Build. This version is based on the current content of https://github.com/HL7/fhir-ae-research-backport-ig/ and changes regularly. See the Directory of published versions
Adverse Event (AE) data capture and standardization is a challenge in real world data systems. There are variations in the definitions, workflows, and data requirements across the use cases for patient monitoring, clinical intervention, regulatory safety reporting requirements and clinical trials monitoring.
The Vulcan Adverse Event project was initiated in October 2021 to model the data elements to support the workflow for collecting and reporting serious and non-serious AEs in clinical research to meet the need of regulatory requirements. The goal of this project is to leverage Electronic Health Records (EHR) and other types of real-world data (RWD), such as Personal Health Records (PHR), as electronic source to collect adverse events that occur during clinical trials. In this project, the team leveraged the HL7 FHIR standard identifying gaps between data elements in EHRs, clinical research artifacts. and other existing standards such as:
This IG will allow clinical trial AE representation in systems, including EHRs and PHRs, transmitted via FHIR to other systems; removing the need to transcribe AE data into secondary clinical trial management systems and improving efficiency for health authority reporting. The following workflows are examples to demonstrate the potential use of this AE profile in the clinical research spectrum via FHIR.
The following image shows a notional workflow for collecting and reporting adverse events and serious adverse events (SAE) for pre-market clinical trials. The dotted line represents a future vision of data flow using FHIR which will most likely be covered by a separate MedWatch IG that builds upon this IG.
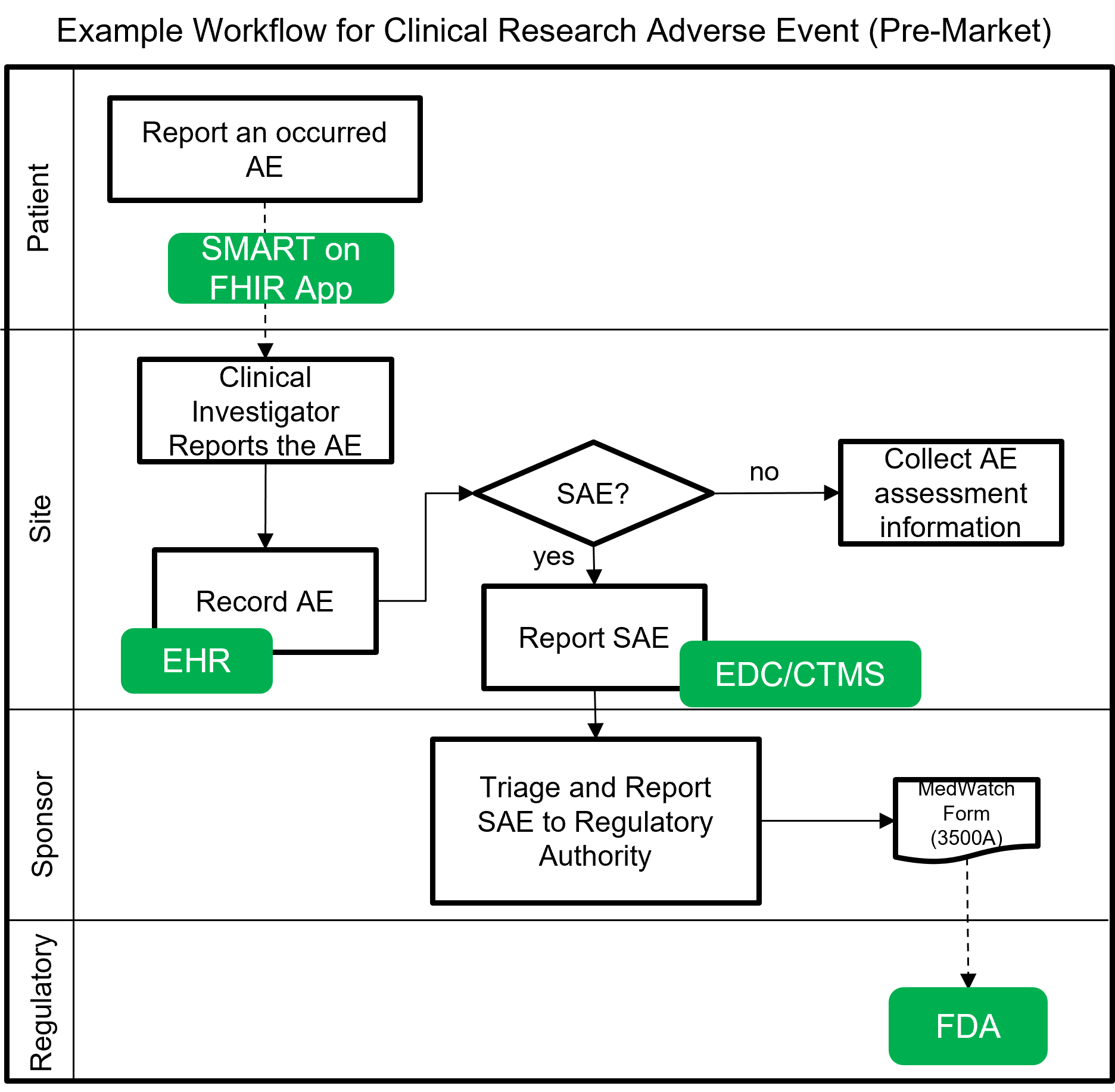
Example scenarios: Pre-market clinical trial reporting of serious adverse events (SAE) using FHIR and utilized as part of clinical decision support actions within a clinical research trial setting.
The following image shows a notional workflow for collecting and reporting adverse events, post-market. The dotted line represents a future vision of data flow using FHIR which will most likely be covered by a separate MedWatch IG that builds upon this IG.
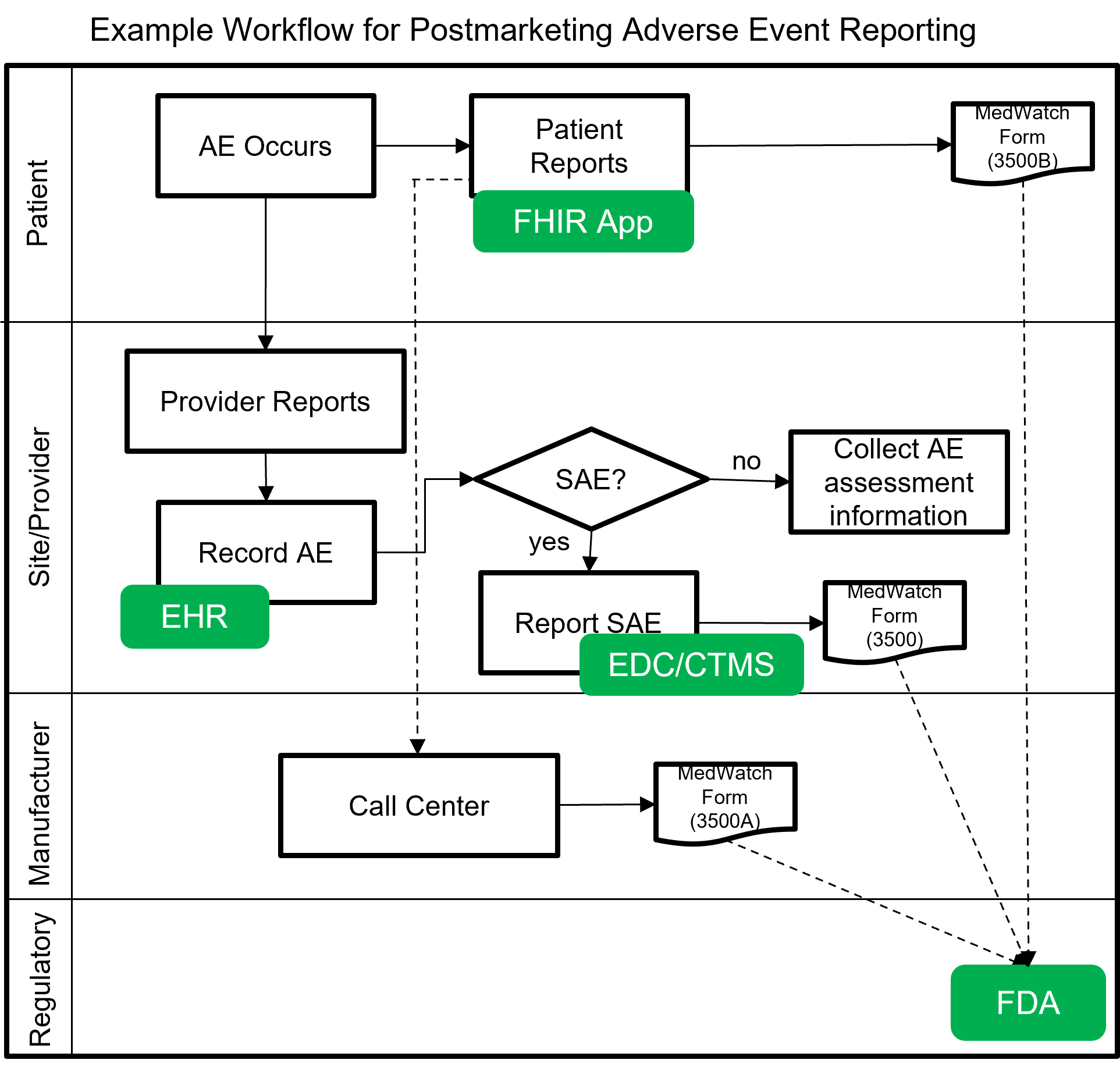
Example scenarios: Postmarketing adverse event reporting using FHIR for safety surveillance.
IG © 2020+ HL7 International / Biomedical Research and Regulation. Package hl7.fhir.uv.ae-research-backport-ig#1.0.1 based on FHIR 4.0.1. Generated 2024-04-30
Links: Table of Contents |
QA Report
| Version History |
 |
Propose a change
|
Propose a change